While the research focus of the Sinz lab is on advancing the cross-linking mass spectrometry (XL-MS) approach for structural and functional proteomics applications, other techniques in structural mass spectrometry are also being increasingly used. These techniques comprise native mass spectrometry, hydrogen-deuterium exchange (HDX), ion mobility-mass spectrometry, and footprinting methods.
1. Protein Structure Analysis by Mass Spectrometry
Until a few years ago, and before the advent of system-wide approaches, analysis by MS-based methods usually required the purification of the protein or protein complex of interest. Notably, the degree to which the sample is purified dictates the quality of the data that are obtained during subsequent MS analysis. For obtaining large yields based on recombinant expression systems are usually applied. As host cells, both prokaryotic and eukaryotic systems can be exploited nowadays and expression vectors for many host cells are commercially available. Highest yields of recombinant protein (up to 80 % of total cellular protein, depending on protein and expression vector) can be retrieved from the Gram-negative bacterium Escherichia coli as host system. However, post-translational modifications and associated interacting protein partners are lost unless they are co-expressed. To meet the latter challenge, various biochemical approaches have been developed for the isolation of endogenous protein complexes directly from cells. For example, one of the subunits or components of the protein complex can be labeled via genetic engineering with a N- or C-terminal peptide tag (FLAG-, His-, GST-, TAP-tag). The high-affinity tag interacts with another component that allows the isolation of the entire protein complex by affinity chromatography under conditions that maintain the integrity of the protein complex 23. In the last years, adoption of these approaches has caused a significant shift in the development of MS-based methods towards the study of highly complex protein mixtures, including system-wide studies in cellulo and in vivo 24.
In general, MS measures the mass-to-charge ratio (m/z) of an ion. Analytes, such as peptides, proteins or medium- to low-molecular weight molecules, are ionized in a first step and then transferred into the gas phase. The ions are subsequently separated, depending on their m/z ratios, in the mass analyzer and ultimately reach the detector. One of the main ways to ionize peptides and proteins is electrospray ionization (ESI) 25, which has evolved as the method of choice for protein analysis. A complementary soft ionization technique, employing matrix-assisted laser desorption/ionization (MALDI), can also be used, although its application has lost some of its importance for protein analysis during the last few years 26,27,28,29. Hybrid instruments that are based on a combination of two or more mass analyzers allow performing tandem MS (MS/MS) experiments 30. In this type of analysis, a specific m/z ratio is isolated from the rest of the ions and activated in a collision cell to induce dissociation of the molecule into smaller components. The resulting fragment ions are then interrogated in a second mass analyzer. Several types of mass analyzers, including time-of-flight (TOF), quadrupole, orbitrap, are currently available, as are various dissociation methods, e.g., collision-induced dissociation (CID), electron-transfer dissociation (ETD), electron-capture dissociation (ECD), surface-induced dissociation (SID), and ultraviolet-photodissociation (UV-PD) 31,32. As a result, a number of configurations of mass spectrometers, each of which vary in their capabilities and limitations, can be designed for different purposes and scientific needs.
2. Native MS
Intact proteins and macromolecular complexes can be studied under non-denaturing conditions in the gas phase of a mass spectrometer through gentle ionization, such as ESI 33. This technique has been termed “native MS” and can provide information on the stoichiometry of non-covalent partners including proteins and non-proteinaceous molecules 34. Native MS provides information on the preserved overall architecture and the conformational changes of complexes 35,36. When used in combination with high-resolution mass spectrometers, native MS has become a valuable technique in the structural proteomics toolbox 37, with one major benefit being able to capturing all coexisting subpopulations in a single spectrum. This enables analysis of different conformational states within multimeric protein complexes that are prompted by ligand bonding and aids elucidation of allosteric mechanisms 38.
Over the last decade, native MS has become a powerful tool for the study of challenging membrane protein complexes and their associated lipids that stabilize a complex 39 or are required for its formation or regulation 40,41. The first structural study with native MS of a membrane complex preserving bound ligands was performed by protecting the complex in micelles using N-dodecyl-ß-D-maltoside as detergent 42. Although new detergents, which are compatible with native MS, are continuously under development 43, alternative approaches are currently being investigated to mimic the phospholipid bilayer of cell membranes, such as nanodiscs 44 and more recently lipodisqs 45. A significant recent advance has involved the analysis of membrane complexes ejected from lipid vesicles obtained from endogenous biological membranes 46.
Despite the great potential of native MS for investigating intact proteins, sample preparation is often tedious as it requires purified protein(s), often involving multiple biochemical isolation steps prior to the last buffer-exchange into the volatile buffer needed for native MS 47. Recently, however, a novel approach has been developed that alllows a more direct analysis of overexpressed proteins from crude extract with minimal sample handling 47,48. This approach is still in its infancy and currently depends on the system used for protein production. However, it will be valuable for the study of proteins at the endogenous level in cells, also taking into account naturally occurring post-translational modifications.
3. Ion Mobility MS
Ion mobility (IM) spectroscopy measures the mobility of an ion in a buffer gas present in a drift tube under the influence of a weak electric field. The time required for an ion to pass through the tube depends on its collision frequency with the drift gas molecules, and thus correlates with its size and shape. This correlation allows derivation of the rotationally averaged collisional cross section (Ω, CCS) of the ion through a mathematical equation 49,50. The CCS derived from the experiment is used to infer structural information, with the deduced information being dependent on the method used 51. Consequently, in order to obtain comparable measurements on different IM platforms, calibration and harmonization of data reporting are important 49,52. IM-MS has served for analytical purposes for more than 50 years, conferring an additional ion separation dimension with gas-phase mobility separation in the IM and m/z determination in the MS 49. During the past 15 years IM-MS has experienced an exponential increase in interest in several scientific fields, particularly, for “omics” studies 53. The rapid spread of IM-MS began with the introduction of the first commercially available mass spectrometer coupled with IM separation 49 and evolved with the subsequent development of different IM technologies, such as drift tube (DTIMS), traveling wave (TWIMS), trapped (TIMS), and field asymmetric ion mobility spectroscopy (FAIMS) 37. These IM platforms differ with respect to gas flow direction, electrostatic and to the electrodynamic fields applied 54. It should be noted that the FAIMS technique, which relies on ion separation field strength, does not allow a reliable CCS measurement 55. The most successful coupling of IM is in combination with nano-ESI and native MS 56, which exploits the combined separation of isomeric ions of different sizes and shapes of all intact coexisting complexes based on their distinct CCS and m/z ratios.
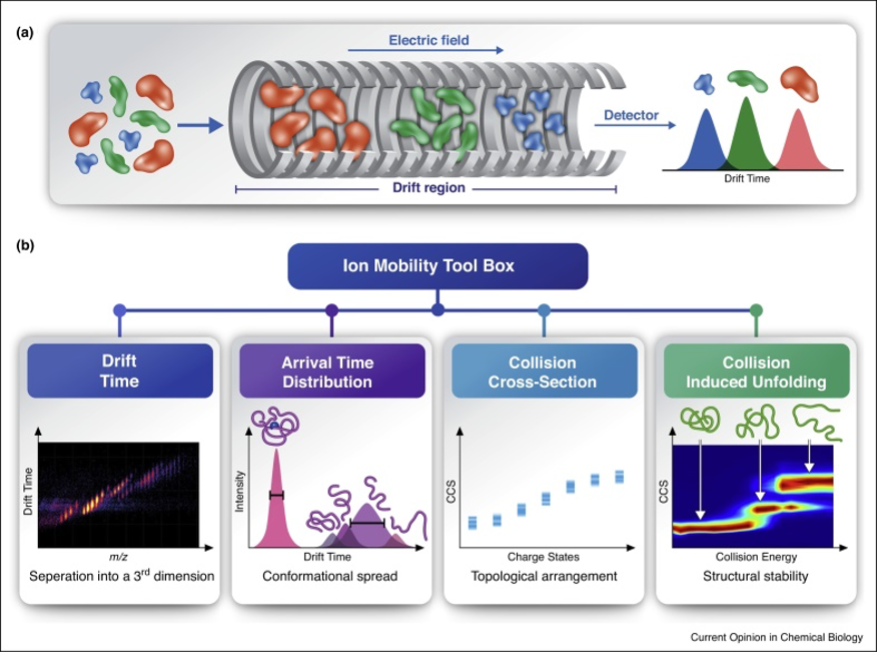
IM-MS of intact protein complexes with its possibility of inducing gas-phase dissociation of the multimeric complex provides important insights into stoichiometry, size, and architecture of protein assemblies (Figure 1) 10,57,58,59.
Figure 1: Structural characterization of protein complexes by IM-MS.
(a) Ion mobility is a method that measures the time it takes for ions to travel
through a gas-filled chamber, across which a weak electric field is applied. In
the drift tube, ions experience two counter forces of electrostatic pulling through
the cell and collisions with the buffer gas, which delay their movement. Ions with a
larger surface area will experience more collisions with the buffer gas and as a
result, will take longer to traverse the drift tube. In comparison, smaller, more
compact ions of the same molecular mass and charge will undergo fewer collisions
with the buffer gas, and hence will display greater mobility and a shorter drift time.
(b) IM-MS measurements enable the extraction of information on various structural
properties. For example, mobility-based separation adds a third dimension to native
MS analyses, and resolves heterogeneous samples that contain closely related species.
Measurements of arrival time distributions reflect the conformational range of the
examined species, and can be converted to CCS values. When measured over a range of
collision energies, as is characteristic of CIU protocols, CCS can reflect the stabilities
and intermediate folding states of the different protein species while they undergo in vacuo
unfolding. Figure from Ben-Nissan et al 57.
A more recent approach shows that the resolution of protein isoforms with small differences in their structural arrangement can be rapidly achieved using their characteristic unfolding behavior and stability after collisional activation before the drift tube 60. Increasing collisional energies are applied to the ion of interest and different CCS values of the activated ion are recorded and plotted as a function of the applied voltage, resulting in a specific collision-induced unfolding (CIU) fingerprint of the molecule 61.
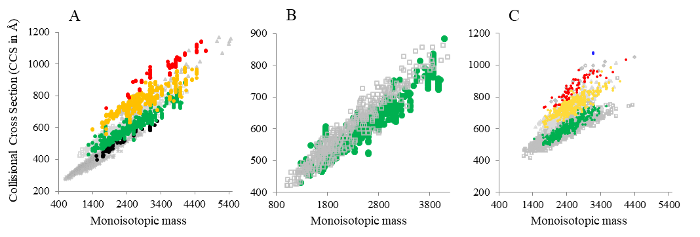
Just recently, IM-MS has been coupled with XL-MS (Figure 2). This combination allows a deeper and more sensitive identification of cross-links 62,63,64, especially in a highly complex environment, paving the way for another successful IM combination.
Figure 2: Distributions of monoisotopic mass versus CCS. (A) Cross-linked and digested E. coli ribosome, (B) zoom-in view on 3+ charge states of cross-linked and linear peptides from the E. coli ribosome, and (C) digested HEK293T cell nuclear lysate. Cross-linked species (intra-, inter-peptide, and “dead-end” cross-links) are color-coded based on their their charge states (• 2+, • 3+, • 4+, • 5+, • 6+); linear peptides are shown in grey (⨯ 2+, ⧠ 3+, △ 4+, ◊ 5+, ○ 6+). Figure from Ihling et al. 62.
4. Hydrogen-deuterium exchange MS
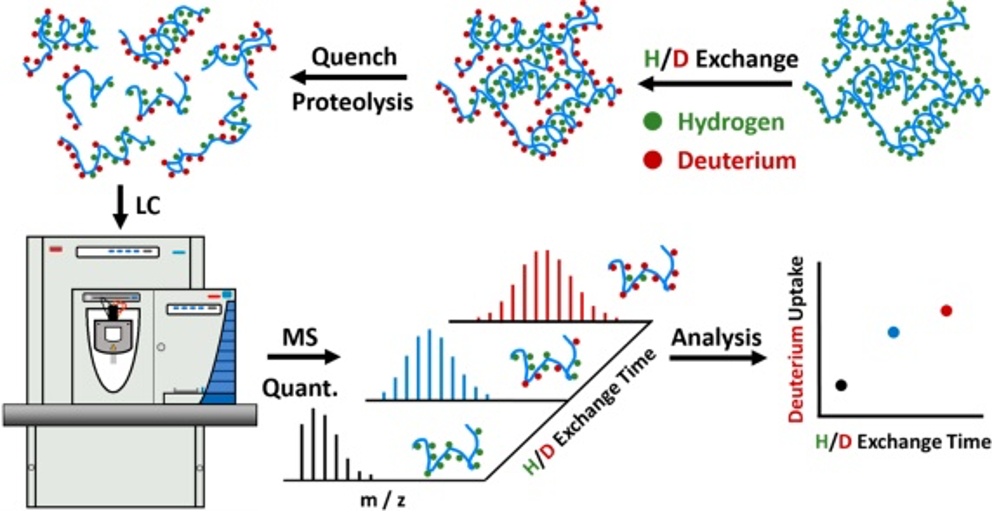
Hydrogen-deuterium exchange combined with mass spectrometry (HDX-MS) presents the most popular protein footprinting method so far developed. HDX monitors the rates of exchanging hydrogen by deuterium atoms at the amide backbone of proteins from solvent D2O in solution 65,66. These exchanges reflect the solvent-accessible surface area (SASA) and are recorded by a mass spectrometer as a function of time. Thus, HDX-MS provides information on solvent accessibility, secondary and tertiary structures, conformational changes, protein states and binding interfaces 67,68. HDX-MS is able to capture protein dynamics and allosteric regulation in solution of a protein or a protein complex, and as such is a local sensor for conformational changes at single amino acid resolution 69 (Figure 3).
Figure 3: Schematic illustration of a bottom-up HDX workflow. Green and red dots in the protein structure indicate hydrogen and deuterium atoms in the peptide bonds, respectively. Figure from Liu et al 70.
Although HDX-MS has been used for 30 years 71, the development of mass analyzers and data analysis software - which remains the bottleneck - have only recently rendered HDX-MS a popular tool among the structural proteomics methods72 enabling studies ranging from single proteins 71 up to viral particles 73. Some drawbacks of HDX are its propensity for back-exchange reactions and D/H scrambling effects 69. In order to quench deuterium exchange and reduce the back-exchange after labeling, the pH value has to be adjusted to ~2.5 and the temperature has to be lowered to ~0°C 74. However, these conditions cause suboptimal efficiency of downstream steps, such as disulfide bonds reduction and protease digestion. The digestion of glycosylated proteins is especially impaired, and among those proteins, the most studied are antibodies and their interactions with ligands 75. To overcome these weaknesses, novel approaches are being evaluated, implemented and integrated in order to be used online and in an automated way, e.g. electrochemical reduction76 and by use of different deglycosylation enzymes that are active at low pH 77.
Meanwhile, the hydrogen-scrambling effect that is mainly instrument-dependent, has been addressed with improvements in mass spectrometer components and electron-based fragmentation methods 78.
In the last years, use of HDX-MS has experienced widespread application in drug development 79 where epitope mapping in antibodies 80, conformational changes in important targets proteins, such as G-protein-coupled receptors in the presence of ligands 81, or biopharmaceutical comparability are critical aspects 82.
5. Chemical Footprinting
Different covalent protein labeling methods such as HDX (see above), covalent labeling, covalent protein painting (CPP), protein painting, hydroxyl radical footprinting (HRFP), and fast photochemical oxidation of proteins (FPOP) are united under the umbrella of MS-based chemical footprinting approaches70. Chemical footprinting relies on covalent labeling of a protein or protein complex in solution to cause a mass shift by the label, which is then detected by MS. The chemical modifications allow probing the structure of proteins, conformational changes, and binding sites in a complex 70,83,84.
Covalent Labeling relies on a covalent chemical reaction targetting side chains of specific amino acids, such as lysines (containing amine groups) or cysteines (containing thiol groups) 70,85. The labeling reagents are chemical compounds that, in contrast to free radicals, create mass tags on the molecules, which are identified in the subsequent MS analysis 70.
Covalent labeling combined with MS has a lot in common with XL-MS and the two techniques complement each other perfectly: XL-MS delivers distance constraints between two amino acids, simultaneously giving information on the orientation of interacting proteins (see below), while covalent labeling indicates the protected areas of the proteins involved in the interaction 85,86.
Covalent labeling has proven highly useful for elucidating the structures of membrane proteins and their interactions87, as well as determining protein-ligand affinities 88. Moreover, it has also been applied to determine the energetic barrier of a structural change that initiates protein fibril formation 89.
Protein Painting employs small-molecules that bind non-covalently to the surface of proteins and therefore block tryptic cleavage sites. However, a limitation of this method is that only peptides involved in the binding interface to another protein, or a small ligand, will be protected from being labeled and will appear as tryptic peptides in subsequent MS analysis 90. The major advantage of covalent protein painting (CPP) is that it directly determines the specific functional groups in proteins, i.e., lysines or cysteines, which are available for a covalent modification on the surface of a protein. CPP is therefore synonymous with covalent labeling. Recently, CPP has been employed in a quantitative approach of dimethylating lysine residues in proteins with “light” (non-deuterated) and “heavy” (deuterated) reagent to detect structural changes of proteins in human postmortem brain samples of patients with neurodegenerative diseases 91.
Hydroxyl Radical Protein Footprinting (HRPF) is an irreversible, non-specific oxidative reaction that relies on hydroxyl radicals. The solvent-exposed side chains of the protein complex react on the order of milliseconds with the radicals providing direct information on the topology of the protein 70. The modification the amino acid side chain undergoes differs for the individual amino acids 92. There are several methods that can be employed to produce hydroxyl radicals, with the most commonly used for protein footprinting being Fenton and Fenton-like chemistry 93, synchrotron water radiolysis 94,93, and laser photolysis of hydrogen peroxide 95. Recently, two novel benchtop approaches have been proposed: Plasma-induced modification of biomolecules (PLIMB) 96; and the Flash OXidation (FOX) Protein Footprinting System 97, which will broaden the array of HRPF tools available for protein structural studies.
Fast Photochemical Oxidation of Proteins (FPOP) is a rapid HRPF method, taking advantage of covalent protein labeling, which occurs on the microsecond timescale. Radicals are produced from H2O2 through photolysis induced by a laser at 248 nm in a microflow system 95,98. FPOP has been applied in a series of investigations, for example, to study protein folding kinetics in the microsecond range 99, epitope mapping 100, refolding processes of viral fusion proteins 101, and characterizing membrane proteins in their native environment 102. This broad range of applications makes FPOP a promising and exciting tool for structural proteomics studies 83,84.
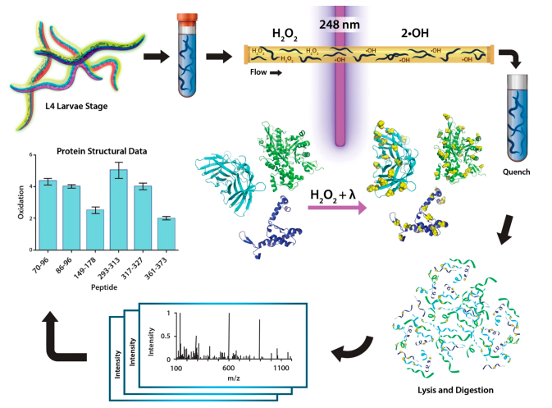
Clearly, one of the most innovative applications of FPOP is in vivo footprinting, where accessible protein regions are labeled directly inside cells without disrupting cellular structures 103. This method has been successfully applied for intact cells and even for living organisms, such as Caenorhabditis elegans104 (Figure 4). In particular, in vivo FPOP has benefitted from a combination with microflow systems98 or static platforms 105.
Figure 4: FPOP workflow for deriving structural information on proteins in live C. elegans larvae. Worms are grown to the fourth larval stage (L4) on nematode growth media plates. For IV-FPOP, worms are passed through a 250-μm fused silica capillary in the presence of H2O2, and radicals are generated using a 248 nm wavelength excimer laser. Immediately after irradiation, excess H2O2, and radicals are quenched, worms are lysed, the protein extract is digested and prepared for mass spectrometry analysis, and the extent of FPOP modifications is calculated for the proteins of interest. Figure from Espino and Jones 104.