The research interest of our group is the advancement of the chemical cross-linking MS (XL-MS) strategy for elucidating protein 3D-structures and for mapping protein-protein interactions. We are currently exploring the development and application of novel MS-cleavable cross-linkers as well as the incorporation of photo-reactive amino acids. Analysis of the cross-linked products is performed by the MeroX software developed by Michael Götze. Our lab is equipped with three Orbitrap mass spectrometers (LTQ-Orbitrap XL, Orbitrap Fusion Tribrid, and Q-Exactive Plus, Thermo Fisher Scientific), and two timsTOF Pro mass spectrometers (Bruker Daltonik) all coupled to nano-HPLC systems, a MALDI-TOF/TOF mass spectrometer (Ultraflex III, Bruker Daltonik), and a High-Mass Q-TOF II mass spectrometer (Micromass/MS Vision).
One important focus of our research is on intrinsically disordered proteins (IDPs), which is reflected by the DFG-funded RTG 2467. Specifically, we develop integrated approaches for investigating the topology of proteins through native mass spectrometry and cross-linking/MS, which we apply to the full-length wild-type p53 (complexes with DNA, S100ß, and sirtuins) and alpha-synuclein. Another focus is on studying G-protein coupled receptors (GPCRs) within SFB1423. Here, we investigate the conformational changes of the Y2 receptor upon binding of its ligand, NPY.
XL-MS complements the array of available MS-based techniques for studying structure and dynamics
of the proteins mentioned above. The general principle underlying the XL-MS approach is strikingly
simple; structural information is obtained by inserting a chemical reagent, a cross-linker,
between two functional groups in a protein. The cross-linker has a defined length and is connected
via covalent bonds to functional groups of amino acid side chains, allowing the cross-linked amino
acids to be identified by MS. The positions of the cross-linked amino acids, together with the
cross-linker’s length, impose distance constraints on the 3D-structure of the protein or the protein complex.
By combining these distance constraints and using them as the basis for subsequent computational modeling,
protein 3D-structures can be derived and protein interfaces mapped.
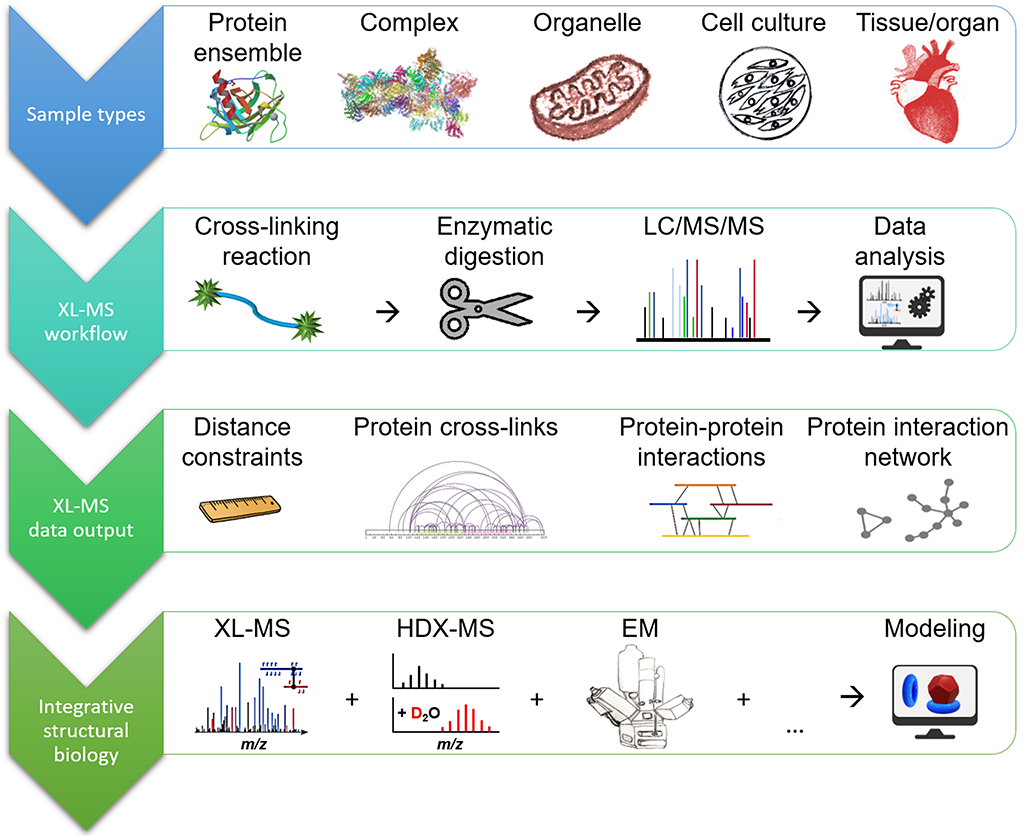
The vast majority of chemical cross-linking studies is performed in a “bottom-up” approach,
whereby after the cross-linking reaction the covalently connected proteins are enzymatically
digested and the resulting peptide mixtures are analyzed by LC/MS/MS (liquid chromatography tandem mass
spectrometry). Typically, ESI is used as ionization technique and orbitrap instruments as mass spectrometers
using collisional activation, for example HCD, to fragment the cross-linked peptides. Both the amino acid
sequences of the connected peptides, as well as the precise cross-link positions can be inferred from the
tandem mass spectra. The mass spectra can be interrogated using customized software tools that allow an
automated assignment of cross-links.
Analysis of cross-linked peptides by MS is the ideal method of choice because it is rapid and requires protein amounts in the femto- to attomole range. In addition, the mass of the protein or the protein complex to be studied is theoretically unlimited. This is due to the enzymatic digestion that is usually performed after the cross-linking reaction, but before conducting the MS analysis at the peptide level. In contrast to most other protein structural methods, it is also possible to study membrane proteins, proteins with post-translational modifications, as well as splice variants. As the cross-linking reactions can be executed in a native environment the cross-links mirror the native conformation of the protein or protein assembly. The large variety of cross-linking reagents with different specificities and spacer distances allows specific tailoring of a XL-MS experiment to the protein system of interest. The technical developments in the XL-MS field are breathtaking, not only with respect to the advances made in MS technology, but also to the development of novel reagents and software tools by many of the groups adopting these approaches. Some of the contributions to these research efforts in the XL-MS field made by our group over the past 20 years are exemplarily depicted in Figure 1.